Despite potential short-cuts, drug development is a lengthy and risky process that culminates in marketing approval. However, just because a drug achieves regulatory approval, does not mean that it will be instantly available to those in need. The complex journey for a pharmaceutical to gain patient access continues.
From discovery to market
The drug discovery process begins when potential therapeutic targets have been identified. Compounds capable of manipulating disease mechanisms to prevent, improve or cure a condition become drug candidates. A pre-clinical phase investigates their therapeutic potential and safety for human testing. Early product development advances in parallel. Regulatory approval, through the Investigational New Drug (IND) application to the US Food and Drug Administration (FDA) or a Clinical Trial Application to European authorities, must be obtained before clinical trials commence. Successfully tested compounds require marketing authorisation before commercial use, in the USA either through a New Drug Application or a Biologics License Application (BLA), in the EU (and Iceland, Norway, Liechtenstein) through a centralised Marketing Authorisation from the European Medicines Agency (EMA) or other interstate processes.
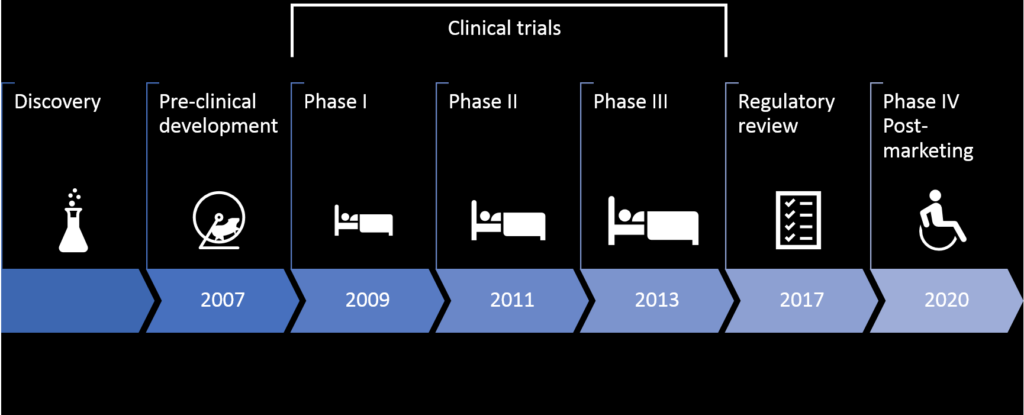
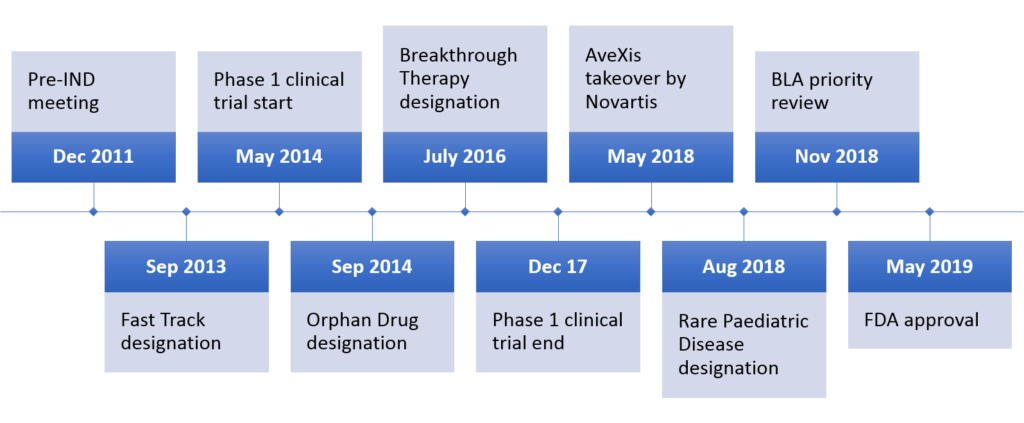
The appraisal process following regulatory approval can delay full market availability by 14 months until reimbursement conditions are clarified. Once on the market in Phase IV, the drug’s therapeutic performance is monitored, for example, to identify long-term side effects. The US process takes 12 years on average (Figure 1); although some drugs can take ‘short-cuts’ through accelerated processes, such as Novartis’ rare disease therapy Zolgensma (Figure 2).
Drug appraisal
Healthcare funding mechanisms can differ greatly between countries, from public or social systems in Europe to heavy reliance on private provisions in the USA. One common component is that marketing approval does not necessarily make a treatment available to patients if payers, such as public healthcare or private insurance providers, refuse to cover its costs. The alternative then are out-of-pocket payments by patients. Thus, when manufacturers’ drug pricing is not aligned with payers’ requirements, an approved cure might become a commercial failure as Glybera‘s case has demonstrated.
Decisive for this hurdle are advisory bodies like the National Institute for Health and Care Excellence (NICE), which makes recommendations to the UK National Health Service (NHS) and is also referred to by private health insurers. Such agencies generally apply some form of Health Technology Assessment (HTA) that weighs up clinical benefits and economic cost-effectiveness. A comparative study based on NICE-appraised drugs from 1999 to 2005 found no difference between the UK and USA in the level of favourable coverage recommendations, with just under 90% of drugs passing the assessments. Conditional coverage and emphasis on cost-effectiveness were more common in the UK, whilst cost-sharing, i.e. required contributions by policyholders, was higher in the USA. In terms of actual coverage, only 36% of total retail pharmaceutical expenditure was covered by US government and compulsory schemes in 2015; together with voluntary health insurance, coverage amounted to 70%. In the UK and across the European Union (EU) 70% and 64%, respectively, of all pharmaceutical costs were covered by public and compulsory schemes in 2016.
In summary, it is apparent that not all drugs that gain regulatory approval also achieve an HTA pass. Even when advisory bodies decide in favour of a drug, healthcare payers may still decide not to go along with that decision or attach conditions. Thus, when big payers refuse to cover such drugs in full or at all, the only way to get access would be out-of-pocket payments by patients. That is, of course, if manufacturers decide to produce the drugs, since it may be uneconomic seeing that sales would be low.
Short-cuts
As indicated above, there are ways to shorten the time to market under certain circumstances. Below are some measures available in the EU and USA that I have come across lately.
Special designation
There are a few special designations for drugs that address specific medical needs. The Zolgensma timeline above (Figure 2) displays some. Once such designations have been granted, the development process can be shortened and/or other benefits are gained by the manufacturer. An interesting case emerged from the current COVID-19 pandemic. In a somewhat unusual move, the FDA granted Gilead’s antiviral remdesivir orphan drug designation, which is generally reserved for rare disease drugs. However, due to public backlash Gilead requested the withdrawal of this designation.
Drug repurposing
Drug repurposing finds alternative uses for marketed drugs or those that have failed in clinical trials. Since a lot of the testing has already been done, getting these drugs out is faster than for brand-new compounds. During 2012–17 nearly 170 drugs underwent repurposing R&D in the US of which 10% were FDA-approved and 72% in clinical trials by 2018/19. Repurposed drugs can make their way to patients via different paths such as the ones outlined below.
Emergency use authorisation
In times of emergency, such as the COVID-19 crisis, unapproved medical products (e.g. drugs, diagnostics, medical devices) may be released, or unapproved uses of approved medical products may be allowed in the absence of adequate approved alternatives. This doesn’t mean that these products/uses are proven to work. They are essentially the best bet to help us through difficult times. This authorisation can be revoked when the required conditions are no longer met. We saw a flurry of emergency use authorisations for COVID-19 diagnostic kits recently because there haven’t been any in existence before. Another topical example is hydroxychloroquine, a drug that has repeatedly made the news during the COVID-19 pandemic. It is an old, cheap malaria drug that has shown some therapeutic promise in early COVID-19 human trials. However, its therapeutic usefulness is unclear at the time of this writing.
By the way, I started tracking news on the development of diagnostics, vaccines and treatments a bit in my monthly biotech review from February 2020 onwards.
Compassionate use
A compassionate use opinion allows unapproved drugs to be used in individual patients outside clinical studies for life-threatening, long-lasting or seriously debilitating illnesses for which there is no adequate approved treatment. However, clinical trials or the marketing authorisation application process must be in progress. Gilead’s remdesivir has been admitted to treat COVID-19 patients through compassionate use in the USA and EU countries. The demand ended up being so high that the company struggled to supply all patients and needed to increase production.
Named-patient basis
Repurposed drugs do not necessarily require new regulatory approval because they can be prescribed ‘off-label’ at the discretion of physicians on an individual patient basis. This is possibly the quickest legal way for a patient to get to use to a drug outside its marketing authorisation as long as the doctor is convinced that it is in the individual’s best interest.
Some additional thoughts
Cost control at all cost?
Whilst overall cost control is important, rigid restrictions on prescriptions can cause physicians professional conflict and frustration. Are treatment decisions to be based on budget constraints or medial judgement? Highly priced novel therapies like Zolgensma are a major drain on budgets. So, should we sacrifice one rare disease patient in order to treat several other patients whose drugs are cheaper? Where would this leave not only medical ethics but also biomedical innovation?
Prices in ads?
Large healthcare payers generally negotiate confidential discounts with pharmaceutical companies. Thus, the usefulness of informing the general public of undiscounted list prices in TV or other advertisements, as has been proposed and rejected, is debatable.
For some background on drug pricing, check out my previous article.
A vulnerable system
Just like other areas of public funding, healthcare is susceptible to macroeconomic events. Currently, we are witnessing an acute strain on healthcare systems worldwide due to the COVID-19 outbreak. We also see enormous levels of investment in COVID-19 R&D and other financial support in relation to the crisis. Quite a few people argue that health systems would not be in such a bad shape, had they not been severely underfunded in the past. It is worth noting that after the 2008 financial crisis, we experienced a chronic decline in healthcare expenditure growth rates in member countries of the Organisation for Economic Cooperation and Development (OECD). With focus on pharmaceuticals, spending dropped by 0.5% annually during 2009-15. This is but one example to demonstrate the far-reaching consequences of excessive financial speculation in a global economy.
References
- AVEXIS, 05/10/2019, 2019-last update, Gene Transfer Clinical Trial for Spinal Muscular Atrophy Type 1 (NCT02122952) [Homepage of ClinicalTrials.gov], [Online]. Available: https://clinicaltrials.gov/ct2/show/NCT02122952 (06/18, 2019).
- BYRNES, A., 2019. Summary Basis for Regulatory Action – ZOLGENSMA. BLA STN#: 125694/0. Silver Spring, MD, USA: US Food and Drug Administration.
- COHEN, J., CAIRNS, C., PAQUETTE, C. and FADEN, L., 2006. Comparing Patient Access to Pharmaceuticals in the UK and US. Applied Health Economics and Health Policy, 5(3), pp. 177-187.
- EMA, 2020. Compassionate use. https://www.ema.europa.eu/en/human-regulatory/research-development/compassionate-use (accessed 11/04/2020)
- FDA, 05/24/2019, 2019a-last update, FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality [Homepage of US Food and Drug Administration], [Online]. Available: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (06/14, 2019).
- FDA, 04/11/2020. About Emergency Use Authorizations (EUAs). https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization (accessed 11/04/2020)
- Florko, Nicholas. 09/07/2019. https://www.statnews.com/2019/07/09/dc-diagnosis-drug-prices-tv-ads/ (accessed 21/08/2019)
- Herper M, 22/03/2020. Gilead pauses access to experimental Covid-19 drug due to ‘overwhelming demand’. https://www.statnews.com/2020/03/22/gilead-suspends-access-to-experimental-covid-19-drug-remdesivir/ (accessed 11/04/2020)
- JACKSON, W.J., PATERSON, A.S., PONG, C.K. and SCARPARO, S., 2014. Cash Limits, Hospital Prescribing and Shrinking Medical Jurisdiction. Financial Accountability & Management, 30(4), pp. 403.
- KESSELHEIM, A.S., AVORN, J. and SARPATWARI, A., 2016. The High Cost of Prescription Drugs in the United States: Origins and Prospects for Reform. JAMA, 316(8), pp. 858-871.
- LEFFEL, E.K., LECLAIRE, R.D. and GHAHRAMANI, P., 2016. Chapter 5 – Regulatory Process in the United States of America, Europe, China, and Japan. In: A. SHAHZAD, ed, Translational Medicine. Boston, USA: Academic Press, pp. 109-133.
- Lupkin S, 24/03/2020. FDA Grants Experimental Coronavirus Drug Benefits For Rare Disease Treatments. https://www.npr.org/sections/health-shots/2020/03/24/821035311/fda-grants-experimental-coronavirus-drug-benefits-for-rare-disease-treatments (accessed 25/03/2020).
- MORENO, S.G. and EPSTEIN, D., 2019. The price of innovation – the role of drug pricing in financing pharmaceutical innovation. A conceptual framework. Journal of Market Access & Health Policy, 7(1), pp. 1583536.
- MULLIN, E., 04/21/2017, 2017-last update, The World’s Most Expensive Medicine Is Being Pulled from the Market [Homepage of MIT Technology Review], [Online]. Available: https://www.technologyreview.com/s/604252/the-worlds-most-expensive-medicine-is-being-pulled-from-the-market (07/05, 2019).
- NOVARTIS, 05/15/2019, 2019-last update, Novartis successfully completes acquisition of AveXis, Inc. [Homepage of Novartis], [Online]. Available: https://www.novartis.com/news/media-releases/novartis-successfully-completes-acquisition-avexis-inc (06/18, 2019).
- O’Day D, 10/04/2020. An Open Letter from our Chairman & CEO. https://www.gilead.com/stories/articles/an-open-letter-from-our-chairman-and-ceo-april-10 (accessed 12/04/2020).
- OECD, 2017. Health at a Glance 2017: OECD Indicators. 9789264280403 (PDF). Paris, France: OECD Publishing.
- OECD and EU, 2018. Health at a Glance: Europe 2018: State of Health in the EU Cycle. 9789264303355 (PDF). Paris; Brussels: OECD Publishing; European Union.
- POLAMREDDY, P. and GATTU, N., 2019. The drug repurposing landscape from 2012 to 2017: evolution, challenges, and possible solutions. Drug Discovery Today, 24(3), pp. 789-795.
- PUSHPAKOM, S., IORIO, F., EYERS, P.A., ESCOTT, K.J., HOPPER, S., WELLS, A., DOIG, A., GUILLIAMS, T., LATIMER, J., MCNAMEE, C., NORRIS, A., SANSEAU, P., CAVALLA, D. and PIRMOHAMED, M., 2018. Drug repurposing: progress, challenges and recommendations. Nature Reviews Drug Discovery, 18, pp. 41-58.
- ROGOWSKI, W.H., HARTZ, S.C. and JOHN, J.H., 2008. Clearing up the hazy road from bench to bedside: A framework for integrating the fourth hurdle into translational medicine. BMC Health Services Research, 8(1), pp. 194.
- Sandler R, 30/03/2020. FDA Authorizes Anti-Malarial Drugs Chloroquine And Hydroxychloroquine For Emergency Coronavirus Treatment. https://www.forbes.com/sites/rachelsandler/2020/03/30/fda-approves-anti-malarial-drugs-chloroquine-and-hydroxychloroquine-for-emergency-coronavirus-treatment (accessed 11/04/2020)
- Silverman E, 25/03/2020. Under intense criticism, Gilead forsakes monopoly status for its experimental Covid-19 drug. https://www.statnews.com/pharmalot/2020/03/25/gilead-covid19-coronavirus-orphan-drug (accessed 7/4/2020)
- Thomas K & Rogers K, 08/07/2019. Judge Blocks Trump Rule Requiring Drug Companies to List Prices in TV Ads. https://www.nytimes.com/2019/07/08/health/drug-prices-tv-ads-trump.html (accessed 12/04/2020).
- WEDA, M., HOEBERT, J., VERVLOET, M., MOLTÓPUIGMARTI, C., DAMEN, N., MARCHANGE, S., LANGEDIJK, J., LISMAN, J. and VANDIJK, L., 2017. Study on off-label use of medicinal products in the European Union. Feb 2017. Brussels: European Union.
- Wirrig, C., 2019. MSc Dissertation: Financing Biomedical Research with Healthcare Derivatives as a Potential Means to Reduce Therapy Prices.
- Wise J, 05/04/2020. US biotech firm donating 1.5 million doses of experimental coronavirus drug. https://thehill.com/homenews/491204-us-biotech-firm-donating-15-million-doses-of-experimental-coronavirus-drug (accessed 11/04/2020)